

standard
行业解决方案
Industry Solutions

通过不断积累与创新的“多元领域、数字智能、质量体系、服务网络”,斯坦德集团可为您提供检验检测、分析研发、计量校准、认证服务、产品质量鉴定、知识产权、标准化/合理化建议等综合型科技服务。
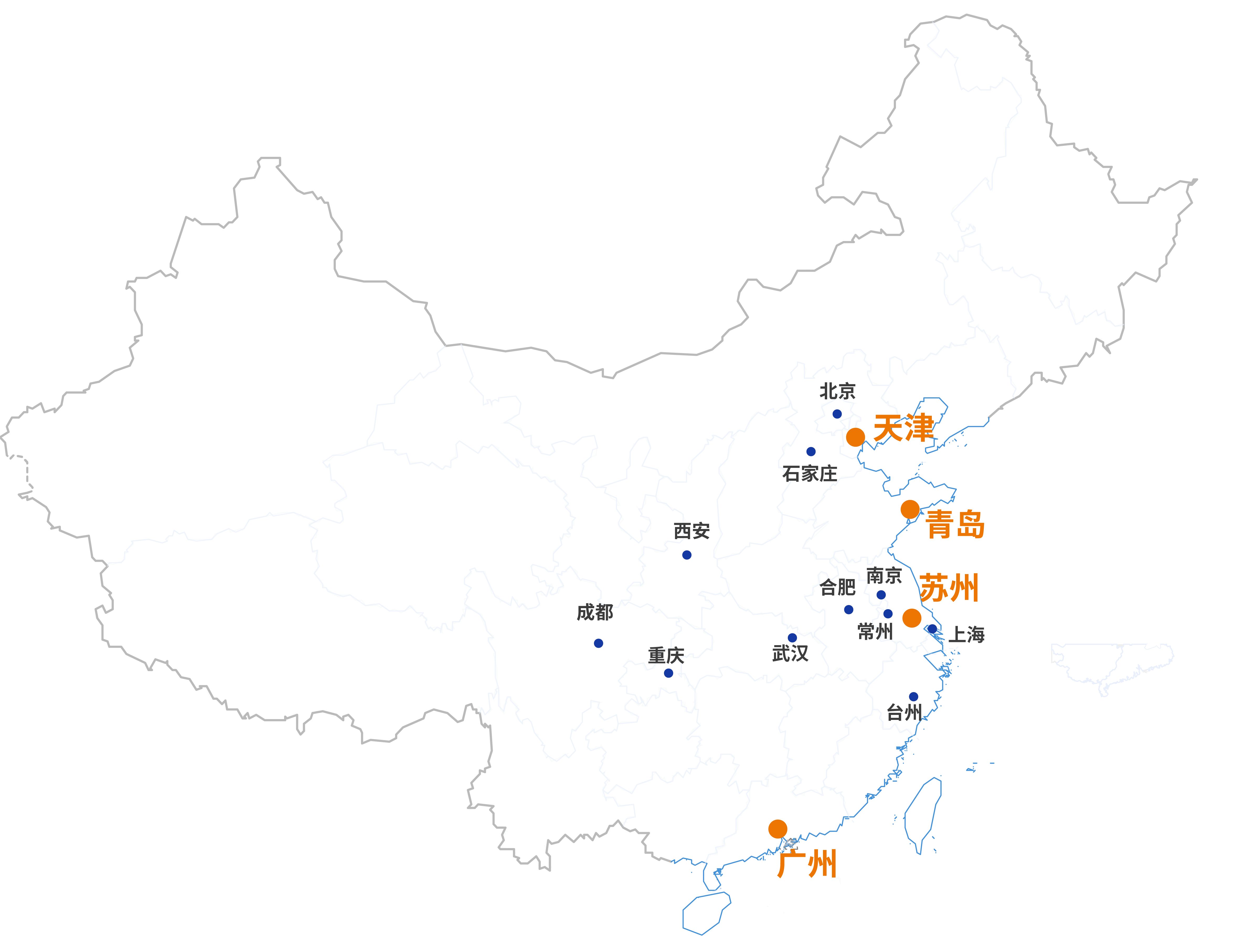
我们的实验室覆盖全国各地区,可迅速响应您的需求,提供多元化的检测研发服务,为品质生活创享信任。
销售联系客户评估并提出方案(约2工作日)
签订合同后实施方案分析测试(约5-7工作日)
出具测试报告并提供售后服务
斯坦德集团,致力于提供多元化的检测研发服务,为品质生活创享信任。